Deriving phase field crystal theory from dynamical density functional theory: consequences of the approximations
Archer, A. J., Ratliff, D.J., Rucklidge, A. M. & Subramanian, P.,
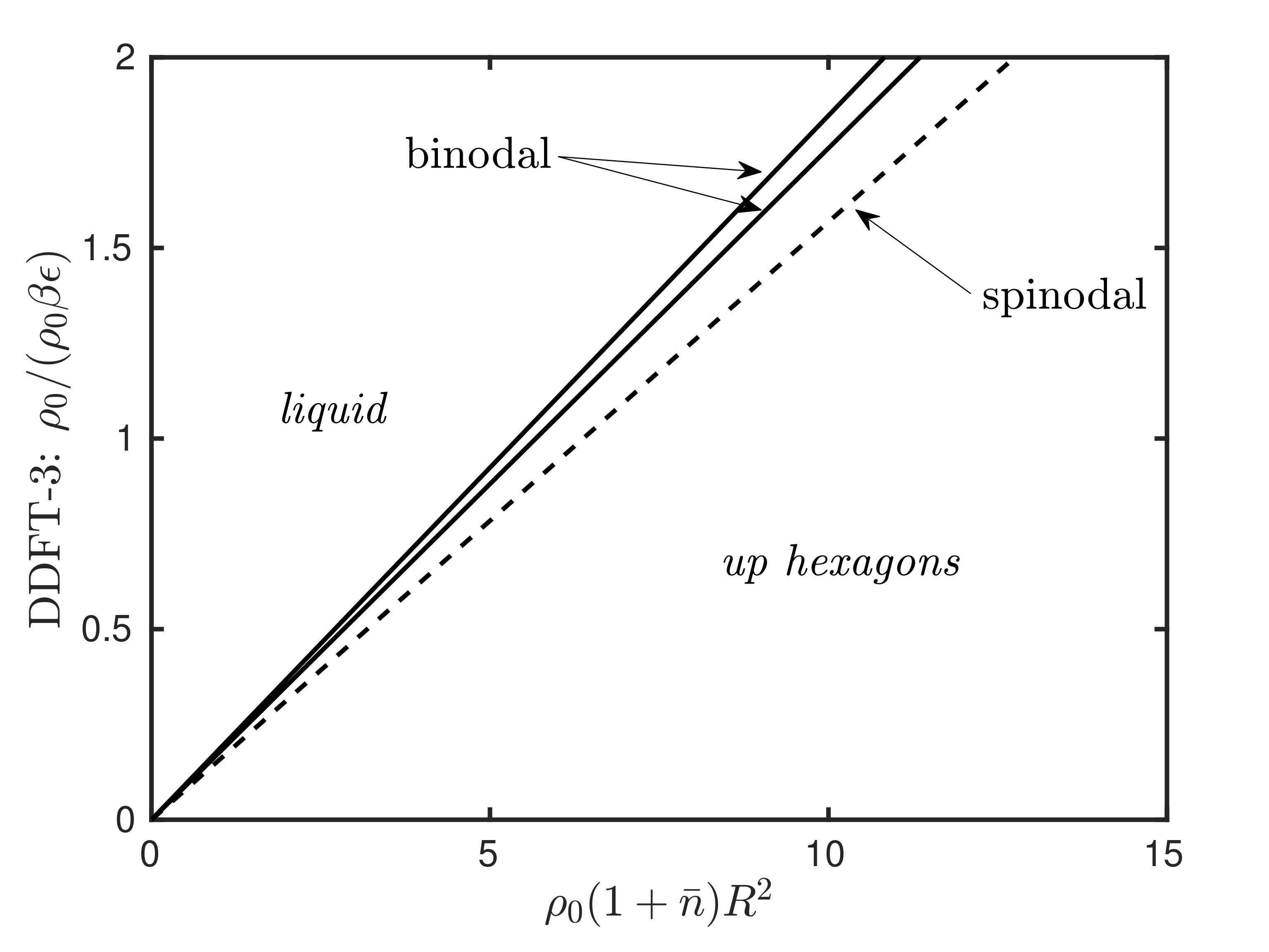
Abstract:
Phase field crystal (PFC) theory is extensively used for modeling the phase behavior, structure, thermodynamics, and other related properties of solids. PFC theory can be derived from dynamical density functional theory (DDFT) via a sequence of approximations. Here, we carefully identify all of these approximations and explain the consequences of each. One approximation that is made in standard derivations is to neglect a term of form ∇⋅[n∇Ln], where n is the scaled density profile and L is a linear operator. We show that this term makes a significant contribution to the stability of the crystal, and that dropping this term from the theory forces another approximation, that of replacing the logarithmic term from the ideal gas contribution to the free energy with its truncated Taylor expansion, to yield a polynomial in n. However, the consequences of doing this are (i) the presence of an additional spinodal in the phase diagram, so the liquid is predicted first to freeze and then to melt again as the density is increased; and (ii) other periodic structures, such as stripes, are erroneously predicted to be thermodynamic equilibrium structures. In general, L consists of a nonlocal convolution involving the pair direct correlation function. A second approximation sometimes made in deriving PFC theory is to replace L with a gradient expansion involving derivatives. We show that this leads to the possibility of the density going to zero, with its logarithm going to −∞ while being balanced by the fourth derivative of the density going to +∞. This subtle singularity leads to solutions failing to exist above a certain value of the average density. We illustrate all of these conclusions with results for a particularly simple model two-dimensional fluid, the generalized exponential model of index 4 (GEM-4), chosen because a DDFT is known to be accurate for this model. The consequences of the subsequent PFC approximations can then be examined. These include the phase diagram being both qualitatively incorrect, in that it has a stripe phase, and quantitatively incorrect (by orders of magnitude) regarding the properties of the crystal phase. Thus, although PFC models are very successful as phenomenological models of crystallization, we find it impossible to derive the PFC as a theory for the (scaled) density distribution when starting from an accurate DDFT, without introducing spurious artifacts. However, we find that making a simple one-mode approximation for the logarithm of the density distribution lnρ(x) rather than for ρ(x) is surprisingly accurate. This approach gives a tantalizing hint that accurate PFC-type theories may instead be derived as theories for the field lnρ(x), rather than for the density profile itself.
Publication details: Physical Review E, 100, 022140, 2019.